Description
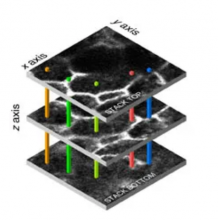
The research goal of this paper was to provide unbiased counts of labeled astrocytes and to estimate the area they cover, further to develop tools for defining the orientation of coupling within astrocyte networks under different stimuli.
In order to count the astrocytes and estimate the area they cover the following steps were used in this software.
Pre-processing: z-project (using max intensity); split channels; subtract background; remove outliers.
Segmentation: adjust threshold and convert to a binary file; Watershed.
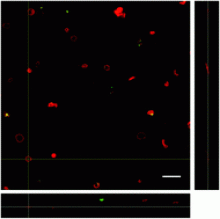
Cell counting: Analyze particles
Measure Astrocytic network area: select a ROI using the polygon tool; set measurements (area); ROI manager -> add the traced polygon; measure.